
Slower and Noisier
Aug. 3, 2020
It seems strange to talk about “quiet” versus “noisy” collisions of neutron stars. But many such impacts form a black hole that swallows all but the gravitational evidence. A series of simulations using PSC’s Bridges platform and other supercomputers by a Penn State scientist suggested that, when the neutron stars’ masses are different enough, the result is far noisier. The model predicts an electromagnetic “bang,” which isn’t present when the merging stars’ masses are similar, that astronomers should be able to detect.
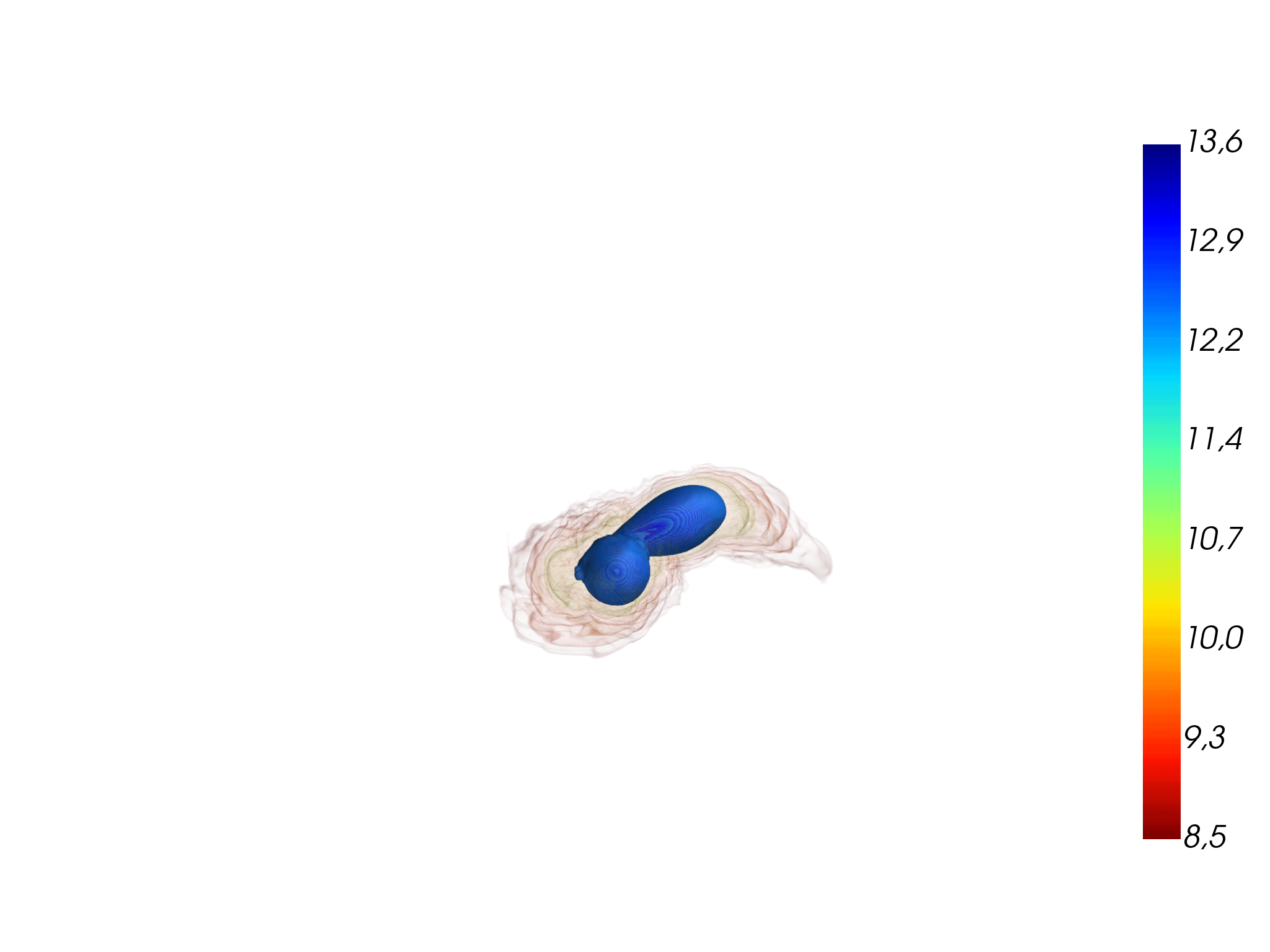
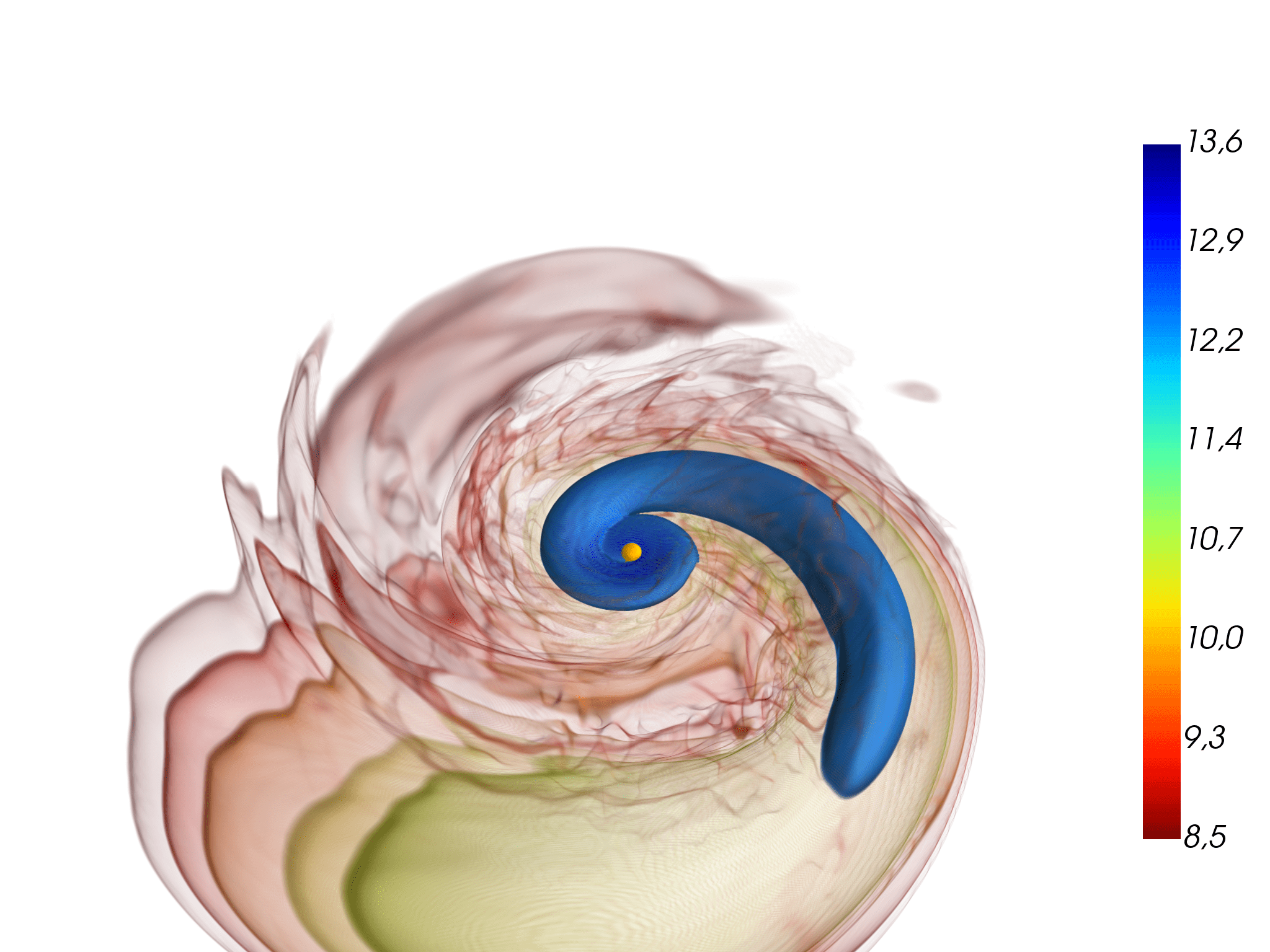
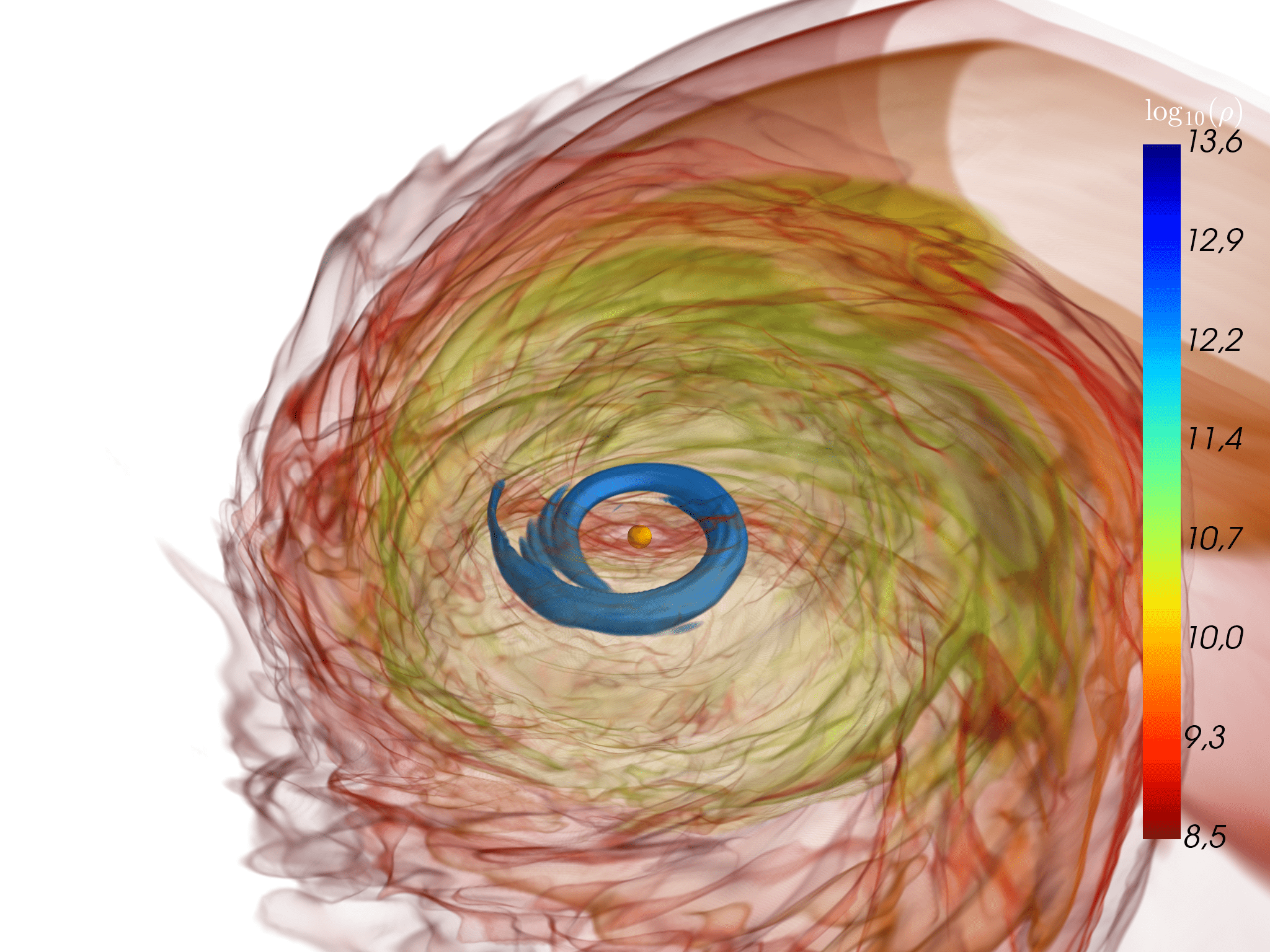
Above: a neutron star is ripped apart by tidal forces from its massive companion in an unequal-mass binary neutron star merger (left). Most of the smaller partner’s mass falls onto the massive star, causing it to collapse and to form a black hole (middle). But some of the material is ejected into space; the rest falls back to form a massive accretion disk around the black hole (right). From Figure 4 in Accretion-induced prompt black hole formation in asymmetric neutron star mergers, dynamical ejecta and kilonova signals. Bernuzzi S et al., Monthly Notices of the Royal Astronomical Society, online June 2020.
Why It’s Important
When two objects roughly the mass of the sun and the size of cities slam together, it seems strange to talk about how “quiet” it is. But for many neutron-star collisions, it is quiet, at least in terms of radiation we can detect. A strong surge of gravitational waves emerges from the impact—now being sensed by gravity-wave detectors such as LIGO, in Hanford, Washington, and Livingston, Louisiana; and Virgo, in Cascina, near Pisa, Italy. But precious little else appears. That’s because the incredibly dense collapsed stars combine to form a black hole, which swallows any of the radiation that could have come out of the merger.
But that’s not the only way it can play out.
“Recently, LIGO announced the discovery of one [merger] event between two stars with possibly very different masses … The main consequence in this scenario is that we expect this very characteristic electromagnetic counterpart [to the gravitational wave signal].”—David Radice, Penn State
After reporting the first detection of a neutron-star merger in 2017, in 2019, the LIGO team reported the second, which they named GW190425. The first of the two collisions was about what astronomers expected, with a total mass of about 2.7 times the mass of our Sun and each of the two neutron stars about equal in mass. But GW190425 was much heavier—a combined mass of around 3.5 Solar masses—and the ratio of the two participants more unequal—possibly as high as 2 to 1.
That may not seem like such a huge difference. But neutron stars can exist only in a narrow range of masses between about 1.2 and 3 times the mass of our Sun. Lighter stellar remnants don’t collapse to form neutron stars and form white dwarfs instead. Heavier objects collapse directly to form black holes. When the difference between the merging stars gets as large as in GW190425, scientists suspected that the merger could be messier—and louder in electromagnetic radiation. Astronomers had detected no such signal from GW190425’s location. But coverage of that area of the sky by conventional telescopes that day wasn’t good enough to rule it out.
David Radice of Penn State, working as member of CoRe, the Computational Relativity International Collaboration, which includes scientists in the U.S., Germany, Italy and Brazil, wanted to better understand the phenomenon of unequal neutron stars colliding, and to predict signatures of such collisions that astronomers could look for. He turned to simulations on PSC’s Bridges supercomputing platform.
How PSC Helped
To run his simulations, Radice needed an unusual combination of computing speed, large memory and flexibility in moving data between memory and computation. That’s partly because scientists know so little about these mergers for certain. To test their ideas, he needed a significant number of computing cores—about 500—running for long periods—weeks—multiple times—about 20, in the current study. Radice employed a number of systems for this work, but the most useful ones he found were PSC’s Bridges and Comet at the San Diego Supercomputer Center, a partner with PSC in the National Science Foundation’s XSEDE network of supercomputing centers and computers.
“There is a lot of uncertainty surrounding the properties of neutron stars. In order to understand them, we have to simulate many possible models to see which ones are compatible with astronomical observations. A single simulation of one model would not tell us much; we need to perform a large number of fairly computationally intensive simulations. We need a combination of high capacity and high capability that only machines like Bridges … can offer. This work would not have been possible without … access to [such] national supercomputing resources.”—David Radice, Penn State
The computations did not disappoint the scientists’ expectations of an electromagnetic bang.
As the two simulated neutron stars spiraled in toward each other, the gravity of the larger star tore its partner apart. That meant that the smaller neutron star didn’t hit its more massive companion all at once. The initial dump of the smaller star’s matter turned the larger into a black hole. But the rest of its matter was too far away for the black hole to capture immediately. Instead, the slower rain of matter into the black hole created a flash of electromagnetic radiation.
The group reported their results online in the journal Monthly Notices of the Royal Astronomical Society in June 2020. Their hope is that the simulated signature they found can be used by astronomers using a combination of gravity-wave and conventional telescopes to detect the paired signals that would herald the breakup of a smaller neutron star merging with a larger one.
You can read their report here.