
Introducing Experimental Data into Molecular Simulations on Bridges Improves Predictions of Metallic Glass Structure
Metallic glass has great strength and flexibility and can be engineered for specific uses. These properties make it useful for medical devices, electrical sensors and even sporting goods. But scientists can’t agree on how the atoms in metallic glasses are arranged. This limits the ability to engineer new metallic glasses for expanded uses. Ohio University scientists employed PSC’s Bridges platform to carry out a new type of molecular simulation on a metallic glass that introduces lab-derived data in the simulation process. The method better agreed with lab results than former simulation methods. It also predicted unexpected properties that may help guide future applications of the material.
Why It’s Important
Amorphous solids—in particular a type called metallic glass—have a strange, not-quite-regular molecular structure. Materials scientists can exploit that irregular structure, “tuning” the properties of metallic-glass materials. This gives these materials many practical uses. Some of these substances serve as strong but flexible components in medical devices. Others can function as pressure-sensitive electrical triggers. They underlie promising advances in nanotechnology. Some are even used in sporting goods.
“There’s an interesting material—a bulk metallic glass—with three different kinds of atoms: palladium, nickel, and phosphorus in a particular composition … It’s very hard to understand its structure. With three kinds of atoms, lots of interactions are possible. What is the physical arrangement of these atoms that really happens in nature? If we knew that, we really could learn something about its properties, thermal and electronic, and how to apply them.”—David Drabold, Ohio University
Scientists can’t agree on exactly how the atoms are arranged in metallic glasses. X-ray or neutron diffraction experiments in the laboratory are usually the highest standard for determining atomic arrangements. But these methods provide inconclusive results because of the materials’ irregular structure. Computer simulations are limited by the massive complexity of the problem. Still, knowing their atomic arrangement would allow scientists to better engineer their properties. Graduate students Bishal Bhattarai (now a postdoctoral fellow at Missouri University of Science and Technology) and Rajendra Thapa, working with Ohio University’s David Drabold, decided to apply a new molecular simulation method called ab initio force enhanced atomic refinement (AIFEAR) to the problem. To run it, they turned to PSC’s National Science Foundation (NSF)-funded Bridges platform.
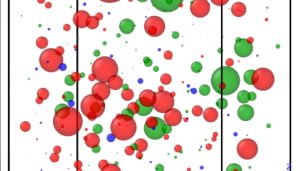
Images showing vibrational activity of different atoms in the Pd40Ni40P20 metallic glass. (Left) A frequency that shows a more common “extended” mode of atoms interacting with each other. Each sphere represents an atom, with smaller spheres showing atoms that are participating in a motion rather than vibrating on their own. (Right) A frequency that shows the vibration of a lone phosphorus atom (blue sphere), rattling inside a “cage” of other atoms without interacting with them much.
How PSC Helped
Amorphous materials have an unusual structure, half-way between crystal and liquid. In crystals, the atoms in array in a strict, repeating pattern. This makes crystals hard but also brittle, since the slightest irregularity is a weak point. Amorphous materials have microscopic, crystal-like bits surrounded by more liquid structure. This gives the materials some of the strength of crystals, but with flexibility as well.
The presence of islands of regular structure within disordered regions makes these materials difficult to study in the lab and difficult to simulate in the computer. To overcome these limitations, the Ohio University scientists used AIFEAR. This new method for simulating the material adjusts the emerging composition with partial information gleaned from laboratory results while finding a good energy minimum—meaning that the atoms’ excess energy reduces. The simulation then cycles in a loop, re-forming the atomic structure with more experimental data until the atoms settle to an energy minimum that can’t be improved upon. Their intention was to improve on the previous simulation method, melt-quench, which optimized its predictions using similar cycles of melting and solidifying the material but without the added laboratory data.
“We start with a random model and refine it by alternating conjugate gradient minimization and experimental data fitting until some convergence is met. As we move on through the AIFEAR steps, we get closer and closer to the experimental information and at the same time get reach a good energy minimum.”—Rajendra Thapa, Ohio University
Bridges proved an important tool for using AIFEAR to study a metallic glass called Pd40Ni40P20. This mixture of palladium, nickel, and phosphorus has proved useful in a number of real-world applications. In addition to offering computing power, Bridges can run a powerful molecular simulation package called VASP that the scientists needed to make AIFEAR work.
The Bridges simulations predicted chemical properties that matched experimental diffraction results better than melt-quench simulations. AIFEAR’s more economical computing also allowed it to simulate a larger system (200 to 300 atoms, as opposed to 100 to 200) than melt-quench, and it ran at least 4 to 5 times quicker than melt-quench. Finally, the NSF-funded computing time on Bridges came without cost, saving the scientists tens of thousands of dollars compared with other systems available. The team published their results in Modeling and Simulation in Materials Science and Engineering in July 2019.
“Bridges is a pretty ideal place to run these simulations … VASP is not universally available, and when I connected with PSC, I was very impressed with the support and help from the staff. They’ve compiled a highly optimized version of the software [which our license allowed us to use]. We were very pleasantly surprised at what was available to us.”—David Drabold, Ohio University
One additional AIFEAR finding, not predicted by earlier melt-quench simulations, was that in some conditions the phosphorus atoms in the material would “rattle” inside a cage of palladium and nickel. This interesting property could give the substance unexpected properties in absorbing and releasing heat that materials scientists might be able to exploit in the future. The team plans more extensive simulation with AIFEAR to better understand and engineer the material.
Read the paper here.